About
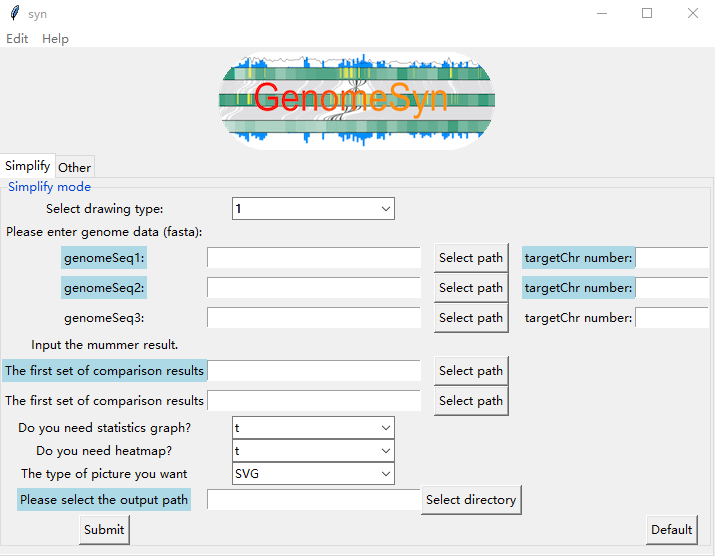
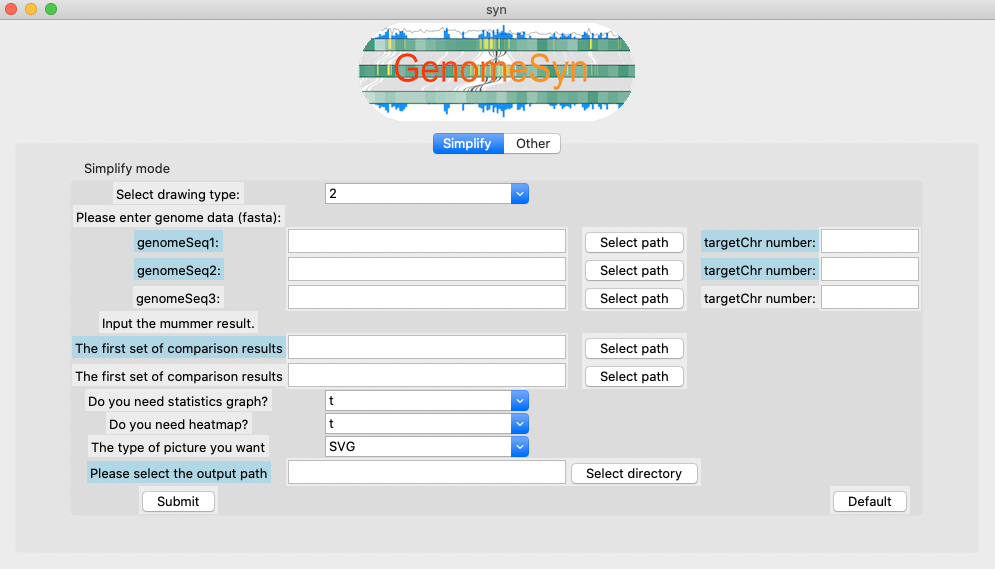
We also prepared a desktop version of GenomeSyn for platforms including Windows, Mac and Linux. The desktop version of GenomeSyn is based on Python, users can click the "Simplify" and "Other" options to select the file to be input. According to the selected target file, click "submit" to show the genomic synteny blocks.
Window Download
Mac Download

Centos Download


README
GenomeSyn - A bioinformatics tool for visualizing genome synteny
================================================================
Development & Fuction
---------------------
GenomeSyn has been tested on multiple platforms including Windows, Mac and CentOS Linux.
The GenomeSyn of the desktop version can achieve the genomic visualization of two to three homologous species,
support the input of the Mummer comparison result.
Publications
------------
See https://cbi.gxu.edu.cn/GenomeSyn/ about for information on citing GenomeSyn.
Installation
------------
There are multiple options on how to get your own copy of GenomeSyn up and running.
See https://cbi.gxu.edu.cn/GenomeSyn/desktop/, click "Download" to download and install.
Double-click the executable synui-cent/mac/win to run the software.
Test:
Users can use example data to test the software GenomeSyn, due to the large amount of example data, the data is not displayed
in this compressed file, so only the download link of the data is provided here:
```
#Download the data for test GenomeSyn
$ https://cbi.gxu.edu.cn/GenomeSyn//download_example/
```
Users can download data from GenomeSyn official website through the above link for testing.
Parameters
----------
-genomeSeq1/
Input the genome1 fasta file to obtain the length of each chromosome in the genome1 (ie query genome).
-genomeSeq2/
Input the genome2 fasta file to obtain the length of each chromosome in the genome2(ie reference genome).
-genomeSeq3/
Input the genome3 fasta file to obtain the length of each chromosome in the genome3(ie query genome2).
-targetChr number/
Input total number of chromosomes in the genome (Total number of items including contig/Scaffold).
-SNP1/
Input the SNP file of genome1, which uses the bed format to map the SNP distribution of genome1.
-SNP2/
Input the SNP file of genome2, which uses the bed format to map the SNP distribution of genome2.
-SNP3/
Input the SNP file of genome3, which uses the bed format to map the SNP distribution of genome3.
-TE1/
Input the TE file of genome1, which uses the bed format to map the TE distribution of genome1.
-TE2/
Input the TE file of genome2, which uses the bed format to map the TE distribution of genome2.
-TE3/
Input the TE file of genome3, which uses the bed format to map the TE distribution of genome3.
-PAV1/
Input the PAV file of genome1, which uses the bed format to map the PAV distribution of genome1.
-PAV2/
Input the PAV file of genome2, which uses the bed format to map the PAV distribution of genome2.
-PAV3/
Input the PAV file of genome3, which uses the bed format to map the PAV distribution of genome3.
-NLR1/
Input the NLR file of genome1, which uses the bed format to map the NLR distribution of genome1.
-NLR2/
Input the NLR file of genome2, which uses the bed format to map the NLR distribution of genome2.
-NLR3/
Input the NLR file of genome3, which uses the bed format to map the NLR distribution of genome3.
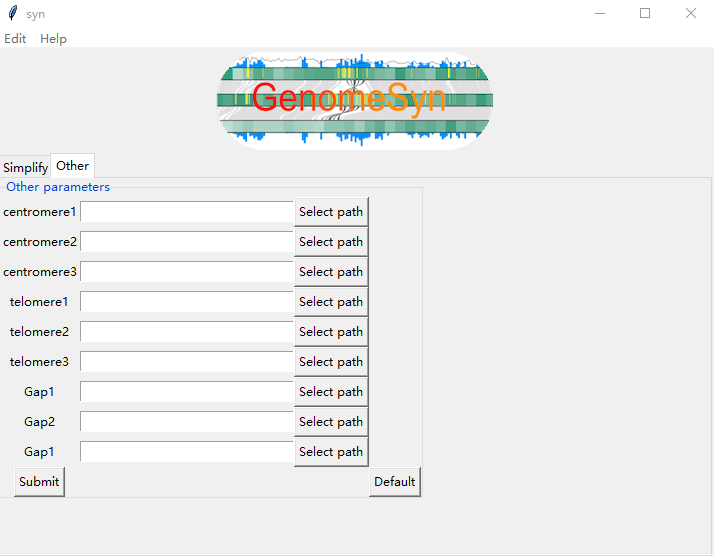
-Telomere1/
Input the telomere position file of genome1, the file uses the bed format, and draw telomere on each chromosome of genome1.
-Telomere2/
Input the telomere position file of genome2, the file uses the bed format, and draw telomere on each chromosome of genome2.
-Telomere3/
Input the telomere position file of genome3, the file uses the bed format, and draw telomere on each chromosome of genome3.
-Centromere1
Input the centromere position file of genome1, the file uses the bed (Browser Extensible Data) format, and draw
centromeres on each chromosome of genome1.
-Centromere2
Input the centromere position file of genome2, the file uses the bed (Browser Extensible Data) format, and draw
centromeres on each chromosome of genome2.
-Centromere3
Input the centromere position file of genome3, the file uses the bed (Browser Extensible Data) format, and draw
centromeres on each chromosome of genome3.
-Gene_density1/
Input the annotation file of genome1, which uses the gff3 format to map the gene density distribution of genome1.
-Gene_density2/
Input the annotation file of genome2, which uses the gff3 format to map the gene density distribution of genome2.
-Gene_density3/
Input the annotation file of genome3, which uses the gff3 format to map the gene density distribution of genome3.
-The first set of comparison results
Input the first MUMmer comparison result file (.coods file).
-The second set of comparison results
Input the second MUMmer comparison result file (.coods file).
-Please select the output path
The path of the output file.
Note
----
1.Please click the "OK" button to confirm the start of the work. According to the server load, your job will take
a few seconds to finish running, please be patient!
2.We have completed the test on existing computers, but we cannot guarantee that it can run well on every computer,
whether it is Windows, Mac or CentOS Linux, if users encounter other problems, users can choose the command line corresponding
to the platform. version runs.
License
-------
GenomeSyn is open source,so users can download the source code and modify the appropriate source code to suit their needs.